
raymolecule is an R package to parse and render molecules in 3D. Rendering is powered by two packages: rayrender package, a pathtracer for R, and rayvertex, a rasterizer for R. raymolecule supports SDF (structure-data file) and PDB (Protein Data Bank) files and can render atom/bond models, ligand overlays, and protein ribbon cartoons.
Installation
You can install the released version of raymolecule from Github:
install.packages("remotes")
remotes::install_github("tylermorganwall/raymolecule")Examples
raymolecule includes several example SDF files for the following molecules: “benzene”, “buckyball”, “caffeine”, “capsaicin”, “cinnemaldehyde”, “geraniol”, “luciferin”, “morphine”, “penicillin”, “pfoa”, “skatole”, “tubocurarine_chloride”. You can get the file path to these example files using the get_example_molecule() function. We pass this path to the read_sdf() file to parse the file and extract the atom coordinates and bond information in a list. raymolecule also includes the ability to fetch molecules from PubChem using the get_molecule() function. The magrittr pipe is automatically imported in the package, so we will use it to pass the output of each function to the input of the next.
Here’s the format of the data:
library(raymolecule)
get_example_molecule("benzene") |>
read_sdf()
#> $atoms
#> x y z type index
#> 1 -1.2131 -0.6884 0e+00 C 1
#> 2 -1.2028 0.7064 1e-04 C 2
#> 3 -0.0103 -1.3948 0e+00 C 3
#> 4 0.0104 1.3948 -1e-04 C 4
#> 5 1.2028 -0.7063 0e+00 C 5
#> 6 1.2131 0.6884 0e+00 C 6
#> 7 -2.1577 -1.2244 0e+00 H 7
#> 8 -2.1393 1.2564 1e-04 H 8
#> 9 -0.0184 -2.4809 -1e-04 H 9
#> 10 0.0184 2.4808 0e+00 H 10
#> 11 2.1394 -1.2563 1e-04 H 11
#> 12 2.1577 1.2245 0e+00 H 12
#>
#> $bonds
#> from to number
#> 1 1 2 2
#> 2 1 3 1
#> 3 1 7 1
#> 4 2 4 1
#> 5 2 8 1
#> 6 3 5 2
#> 7 3 9 1
#> 8 4 6 2
#> 9 4 10 1
#> 10 5 6 1
#> 11 5 11 1
#> 12 6 12 1Alternatively, you can fetch any molecule from PubChem by passing either the molecule name. You can also fetch a molecule using the official compound ID (CID), in case you have a specific molecule with a long name or unique isoform:
str(get_molecule("estradiol"))
#> List of 2
#> $ atoms:'data.frame': 44 obs. of 5 variables:
#> ..$ x : num [1:44] 5.061 -5.857 2.664 1.984 0.515 ...
#> ..$ y : num [1:44] 1.15 0.148 0.584 -0.695 -0.78 ...
#> ..$ z : num [1:44] 0.2674 0.4388 0.1481 -0.3789 0.0353 ...
#> ..$ type : chr [1:44] "O" "O" "C" "C" ...
#> ..$ index: int [1:44] 1 2 3 4 5 6 7 8 9 10 ...
#> $ bonds:'data.frame': 47 obs. of 3 variables:
#> ..$ from : num [1:47] 1 1 2 2 3 3 3 3 4 4 ...
#> ..$ to : num [1:47] 7 40 20 44 4 7 8 13 5 9 ...
#> ..$ number: num [1:47] 1 1 1 1 1 1 1 1 1 1 ...
str(get_molecule(5757)) #this is the CID for estradiol (aka estrogen)
#> List of 2
#> $ atoms:'data.frame': 44 obs. of 5 variables:
#> ..$ x : num [1:44] 5.061 -5.857 2.664 1.984 0.515 ...
#> ..$ y : num [1:44] 1.15 0.148 0.584 -0.695 -0.78 ...
#> ..$ z : num [1:44] 0.2674 0.4388 0.1481 -0.3789 0.0353 ...
#> ..$ type : chr [1:44] "O" "O" "C" "C" ...
#> ..$ index: int [1:44] 1 2 3 4 5 6 7 8 9 10 ...
#> $ bonds:'data.frame': 47 obs. of 3 variables:
#> ..$ from : num [1:47] 1 1 2 2 3 3 3 3 4 4 ...
#> ..$ to : num [1:47] 7 40 20 44 4 7 8 13 5 9 ...
#> ..$ number: num [1:47] 1 1 1 1 1 1 1 1 1 1 ...We can then pass the list from get_example_molecule() |> read_sdf() or from get_molecule() to the generate_full_scene(), generate_atom_scene(), or generate_bond_scene() functions to convert this representation to a raymesh scene. This can then be passed on the render_model() function, which will call rayrender’s render_scene() or rayvertex’s rasterize_scene() functions depending on pathtrace. This function automatically ensures the molecule is centered and in frame, sets up lighting, and can accept arguments to rotate the molecule. For more rendering options, see rayrender::render_scene() and rayvertex::rasterize_scene().
The atom-only and bond-only builders are useful when you want to inspect the simpler pieces of a molecule before rendering a full ball-and-stick model.
#Render atoms only for a small built-in molecule
get_example_molecule("benzene") |>
read_sdf() |>
generate_atom_scene() |>
render_model(width = 800, height = 800, samples = 32)
#> --------------------------Interactive Mode Controls---------------------------
#> W/A/S/D: Horizontal Movement: | Q/Z: Vertical Movement | Up/Down: Adjust FOV | ESC: Close
#> Left/Right: Adjust Aperture | 1/2: Adjust Focal Distance | 3/4: Rotate Environment Light
#> P: Print Camera Info | R: Reset Camera | TAB: Toggle Orbit Mode | E/C: Adjust Step Size
#> K: Save Keyframe | L: Reset Camera to Last Keyframe (if set) | F: Toggle Fast Travel Mode
#> Left Mouse Click: Change Look At (new focal distance) | Right Mouse Click: Change Look At
#> ]/[: Adjust Preview Exposure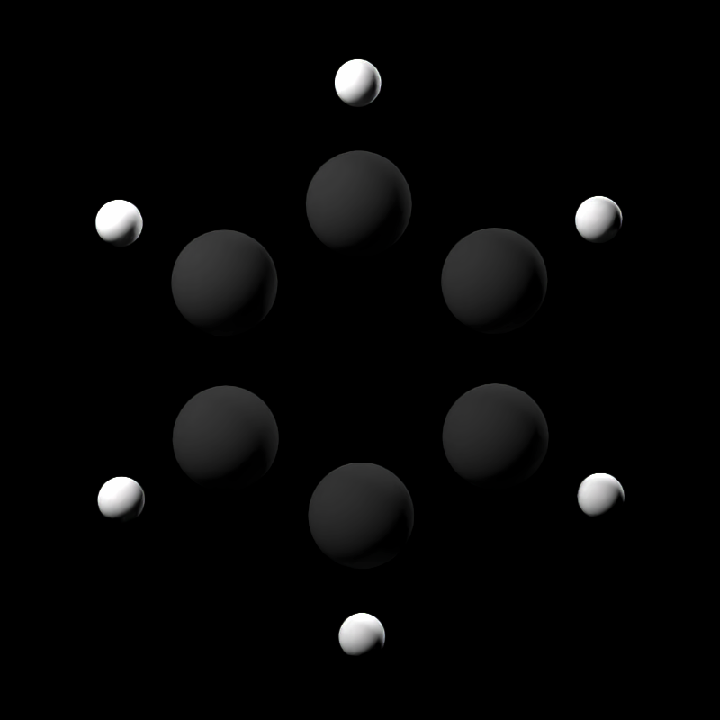
plot of chunk atom-bond-scenes
#Render only the bond geometry for a simple built-in molecule
get_example_molecule("cinnemaldehyde") |>
read_sdf() |>
generate_bond_scene() |>
render_model(width = 800, height = 800, samples = 32)
#> --------------------------Interactive Mode Controls---------------------------
#> W/A/S/D: Horizontal Movement: | Q/Z: Vertical Movement | Up/Down: Adjust FOV | ESC: Close
#> Left/Right: Adjust Aperture | 1/2: Adjust Focal Distance | 3/4: Rotate Environment Light
#> P: Print Camera Info | R: Reset Camera | TAB: Toggle Orbit Mode | E/C: Adjust Step Size
#> K: Save Keyframe | L: Reset Camera to Last Keyframe (if set) | F: Toggle Fast Travel Mode
#> Left Mouse Click: Change Look At (new focal distance) | Right Mouse Click: Change Look At
#> ]/[: Adjust Preview Exposure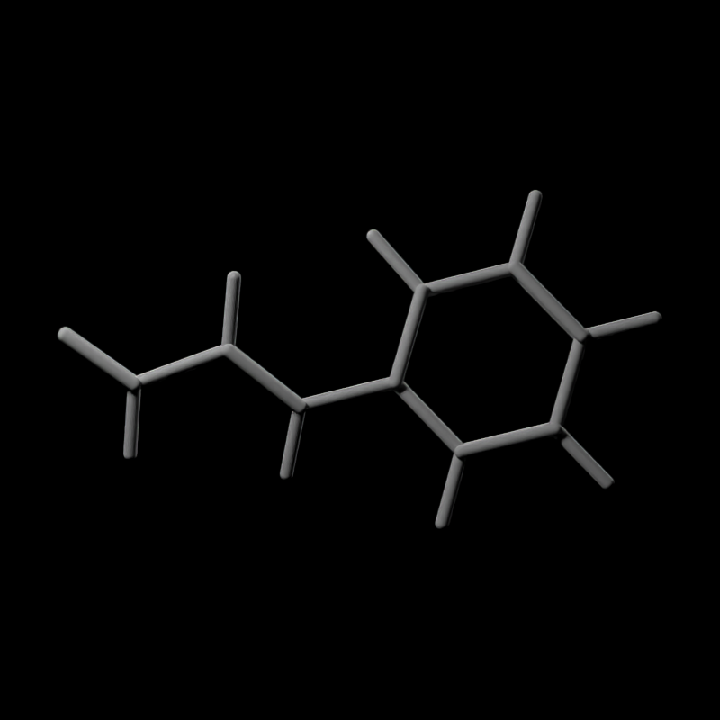
plot of chunk atom-bond-scenes
#Specify a width, height, and number of samples for the image (more samples == less noise)
get_example_molecule("caffeine") |>
read_sdf() |>
generate_full_scene() |>
render_model(width = 800, height = 800, samples = 32)
#> --------------------------Interactive Mode Controls---------------------------
#> W/A/S/D: Horizontal Movement: | Q/Z: Vertical Movement | Up/Down: Adjust FOV | ESC: Close
#> Left/Right: Adjust Aperture | 1/2: Adjust Focal Distance | 3/4: Rotate Environment Light
#> P: Print Camera Info | R: Reset Camera | TAB: Toggle Orbit Mode | E/C: Adjust Step Size
#> K: Save Keyframe | L: Reset Camera to Last Keyframe (if set) | F: Toggle Fast Travel Mode
#> Left Mouse Click: Change Look At (new focal distance) | Right Mouse Click: Change Look At
#> ]/[: Adjust Preview Exposure
plot of chunk unnamed-chunk-5
#Light from both bottom and top
get_example_molecule("cinnemaldehyde") |>
read_sdf() |>
generate_full_scene() |>
render_model(lights = "both", width = 800, height = 800, samples = 32)
#> --------------------------Interactive Mode Controls---------------------------
#> W/A/S/D: Horizontal Movement: | Q/Z: Vertical Movement | Up/Down: Adjust FOV | ESC: Close
#> Left/Right: Adjust Aperture | 1/2: Adjust Focal Distance | 3/4: Rotate Environment Light
#> P: Print Camera Info | R: Reset Camera | TAB: Toggle Orbit Mode | E/C: Adjust Step Size
#> K: Save Keyframe | L: Reset Camera to Last Keyframe (if set) | F: Toggle Fast Travel Mode
#> Left Mouse Click: Change Look At (new focal distance) | Right Mouse Click: Change Look At
#> ]/[: Adjust Preview Exposure
plot of chunk unnamed-chunk-5
#Rotate the molecule and add a non-zero aperture setting to get depth of field effect
get_example_molecule("penicillin") |>
read_sdf() |>
generate_full_scene() |>
render_model(
lights = "both",
width = 800,
height = 800,
samples = 64,
angle = c(0, 30, 0),
aperture = 2,
fov = 22
)
#> --------------------------Interactive Mode Controls---------------------------
#> W/A/S/D: Horizontal Movement: | Q/Z: Vertical Movement | Up/Down: Adjust FOV | ESC: Close
#> Left/Right: Adjust Aperture | 1/2: Adjust Focal Distance | 3/4: Rotate Environment Light
#> P: Print Camera Info | R: Reset Camera | TAB: Toggle Orbit Mode | E/C: Adjust Step Size
#> K: Save Keyframe | L: Reset Camera to Last Keyframe (if set) | F: Toggle Fast Travel Mode
#> Left Mouse Click: Change Look At (new focal distance) | Right Mouse Click: Change Look At
#> ]/[: Adjust Preview Exposure
plot of chunk unnamed-chunk-5
We can use rayvertex to render images much more quickly and noise free, as well as include a toon cel-shading effect.
library(rayvertex)
#Render a basic example with rayvertex
get_example_molecule("tubocurarine_chloride") |>
read_sdf() |>
generate_full_scene() |>
render_model(pathtrace = FALSE, width = 800, height = 800, background = "grey66")
plot of chunk unnamed-chunk-6
#Customize the material with toon shading
shiny_toon_material = material_list(
type = "toon_phong",
toon_levels = 3,
toon_outline_width = 10
)
get_example_molecule("morphine") |>
read_sdf() |>
generate_full_scene(
material_vertex = shiny_toon_material
) |>
render_model(pathtrace = FALSE, width = 800, height = 800, background = "grey66")
plot of chunk unnamed-chunk-6
#Customize the lights with rayvertex
get_example_molecule("skatole") |>
read_sdf() |>
generate_full_scene() |>
render_model(
pathtrace = FALSE,
width = 800,
height = 800,
angle = c(0, 30, 0),
background = "grey66",
lights = directional_light(c(0, 1, 1)) |>
add_light(directional_light(c(0, -1, 0), color = "red"))
)
plot of chunk unnamed-chunk-6
You can turn off lighting in render_model() and customize the rendered output by using the lower-level rayrender or rayvertex APIs directly. If you use rayrender::render_scene()/rayvertex::rasterize_scene() instead of render_model(), you have to set up the camera position and field of view yourself.
library(rayrender)
#>
#> Attaching package: 'rayrender'
#> The following object is masked from 'package:rayvertex':
#>
#> r_obj
buckyball = get_example_molecule("buckyball") |>
read_sdf() |>
generate_full_scene()
#Add custom lighting, with a custom position determined interactively
# in rayrender by moving to the desired position and pressing "P" to get
# the coordinates.
buckyball |>
add_object(sphere(
y = 12,
radius = 3,
material = light(color = "white", intensity = 30)
)) |>
add_object(sphere(
y = -12,
radius = 3,
material = light(color = "red", intensity = 30)
)) |>
add_object(sphere(
x = 12,
radius = 3,
material = light(color = "dodgerblue", intensity = 30)
)) |>
add_object(sphere(
x = -12,
radius = 3,
material = light(color = "orange", intensity = 30)
)) |>
render_model(
lights = "none",
width = 800,
height = 800,
samples = 64,
fov = 10,
aperture = 1
)
#> Error in `rbind()`:
#> ! numbers of columns of arguments do not match
#Generate ground underneath the model and use a light to cast a shadow
generate_ground(depth = -4, material = diffuse(color = "purple")) |>
add_object(buckyball) |>
add_object(sphere(y = 8, z = 12, material = light(intensity = 200))) |>
render_scene(
width = 800,
height = 800,
samples = 64,
aperture = 1,
fov = 20,
lookfrom = c(5.96, 0.99, 29.43),
lookat = c(0.80, 0.26, 3.91)
)
#> Error in `rbind()`:
#> ! numbers of columns of arguments do not matchProtein Data Bank Ribbons
raymolecule can also download PDB structures from RCSB, parse multi-model ensembles, and generate ribbon cartoons. This compact beta-barrel example renders a single PDB structure with UV ribbon coloring and hetero overlays.
pdb_file = download_pdb("4fsp", out_dir = tempdir(), overwrite = TRUE)
read_pdb(pdb_file, verbose = TRUE) |>
generate_ribbon_scene(
color_mode = "uv",
show_hetero_atoms = TRUE,
show_hetero_bonds = TRUE
) |>
render_model(pathtrace = FALSE, width = 800, height = 800, background = "grey12")
#> Read CRYSTAL STRUCTURE OF PSEUDOMONAS AERUGINOSA OCCK11 (OPDR) PDB models [1]
#> PDB ID: 4FSP
#> Experiment: X-RAY DIFFRACTION
#> Parsed: 3135 atoms, 364 residues, 1 chains, 192 bonds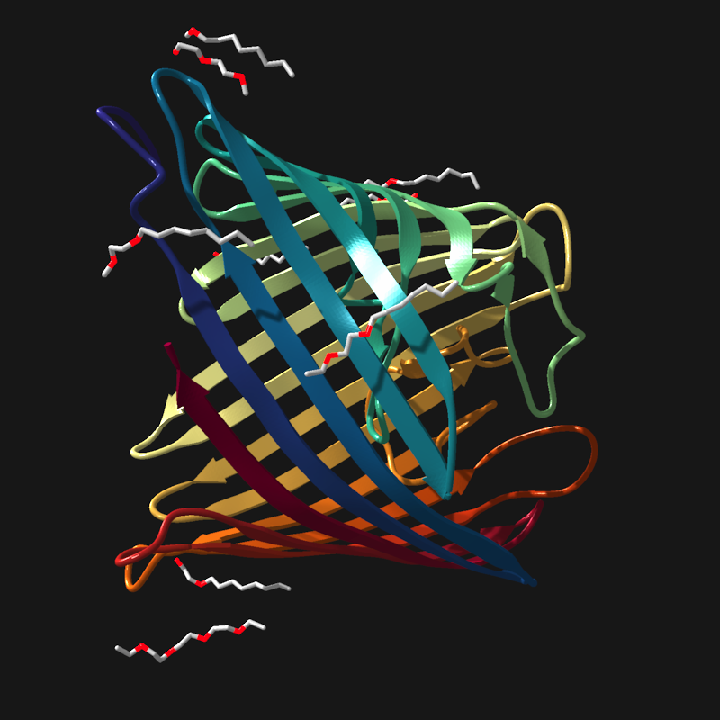
plot of chunk pdb-examples